With age, our skeletal muscles (e.g. muscle of our legs and arms) work less well. In some people, there is a substantial loss of strength and functionality called sarcopenia. This has a knock-on effect on the health of elderly people. If we want to delay skeletal muscle ageing, so that we can stay active and healthy for longer, we need to understand the mechanism behind it.
The spread of a type of mitochondrial mutants, deletions, has been causally implicated in the ageing of skeletal muscle. But how do these mutations expand, replacing normal (wildtype) mitochondrial DNA? One could think that they have a replicative fitness advantage, therefore they outcompete wildtypes. However, no definitive explanation of this supposed faster replication is known for non-replicating cells, despite numerous proposals. We might even expect that these mutants are actively eliminated since they are disadvantageous to the cell.
In a recent PNAS paper, (free version here) we unveil a new evolutionary mechanism, termed stochastic survival of the densest, that accounts for the expansion of mitochondrial deletions in skeletal muscle. We do not assume a replicative advantage for deletions, and even allow the possibility of a higher degradation rate for mutants compared to non-mutants. Our stochastic model predicts a noise-driven clonal wave of advance of mitochondrial mutants, recapitulating experimental observation qualitatively and quantitatively.
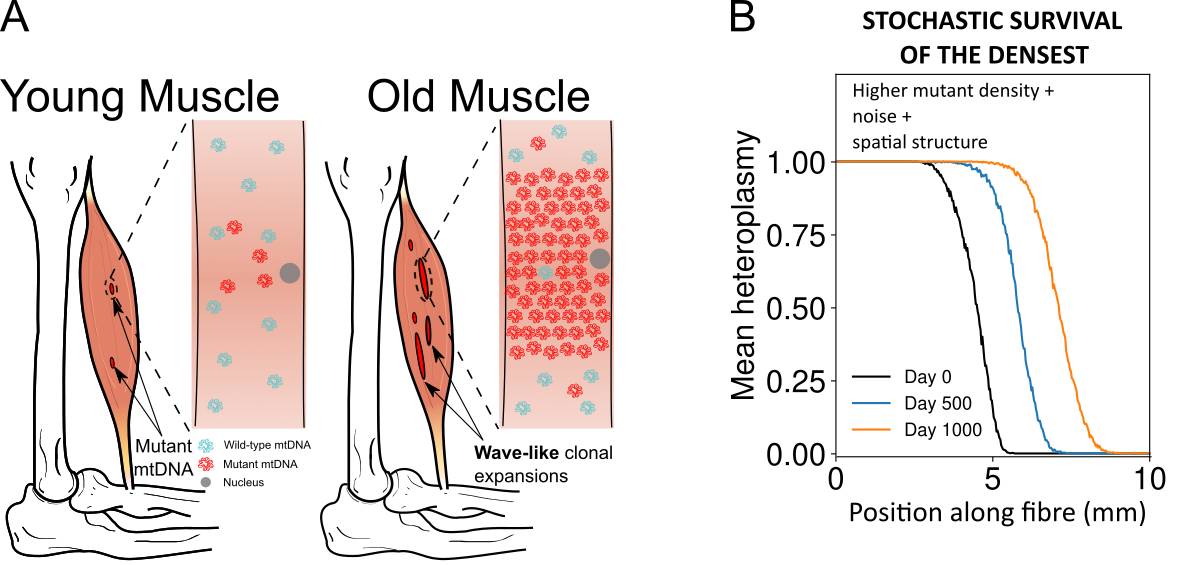
Stochastic survival of the densest accounts for the expansion of mitochondrial DNA mutants in aged skeletal muscle through a noise-driven wave of advance of denser mutants.
A) Dysfunctional mtDNA mutants expand in muscle fibres with age, occupying macroscopic regions where strength and functionality are lost. B) A stochastic model of a spatially extended system predicts a travelling wave of denser mutants (e.g. the mitochondrial deletions in skeletal muscles), according to a novel evolutionary mechanism termed stochastic survival of the densest.
A key reason behind mutants’ expansion is that they can live at higher densities (or carrying capacity) in the muscle fibres. This, together with the stochastic nature of biological processes, gives rise to the surprising wave-like expansion. Remarkably, if you take the noise away, the effect disappears: it is a truly stochastic phenomenon. We are very excited about this new mechanistic understanding: the clonal expansion of deletions has been the subject of intense research for over 30 years. Moreover, this progress allows principled suggestions of therapies that might slow down skeletal muscles ageing.
We are possibly even more excited about the evolutionary implications of our work. We believe that stochastic survival of the densest is a previously unrecognised mechanism of evolution that can account for other counterintuitive phenomena. We essentially showed that, in the presence of noise, a species can take over a system because the more of it there is the faster all species in the system replicate. This conforms to one of the strict definitions of altruism. Therefore, the model can also account for the spread of altruism: a species can win a competition because it is altruistic. The model we use to show the effect is one of the simplest models for populations (generalised stochastic Lotka-Volterra) and we think that the effect is robust, and will be reproduced by a variety of similar models and in a range of geometries/topologies
Mitochondrial deletion mutants are bad for our muscles and our health, but in order to understand (and counteract) their expansion we might consider them an altruistic species that, driven by noise, outcompetes wildtype mitochondria. Ferdinando and Nick